DNA typically is found as a double stranded molecule in which the sequence of nucleotide bases on one strand (for instance, ATCG) has a complementary sequence (TAGC) on the opposite strand. If two complementary DNA strands are mixed together in solution, they tend to join together (hybridize) and form a stable double-stranded molecule by hydrogen bonding. The degree of hybridization between the two strands is dictated by base-pairing rules (for example, A on one strand always binds to T on the other strand) and the specific sequence of bases in the two strands. Two DNA strands with complementary base sequences will bind together tightly, while dissimilar strands will bind together poorly, if at all.
Microarrays exploit this hybridization effect to detect the presence of DNA sequences for specific genes in environmental samples by using short, single-stranded DNA molecules (probes) with known nucleotide sequences. In practice, these probes are attached to a solid surface such as a glass slide or suspended in a gel. After extracting DNA from an environmental sample (and sometimes amplifying the DNA by PCR), the DNA sample is labeled with a fluorescent dye and applied to the array. When hybridization occurs, the labeled DNA that complements the microarray probesShort, defined segments of DNA that are designed to bind with the target gene if found in the environmental sample. The probes are attached to the solid surface of the microarray. are retained on the slide, producing a characteristic fluorescent signal. DNA that does not have a complementary probe on the microarrayDetects and estimates the relative abundances of hundreds to thousands of genes simultaneously. slide does not bind (hybridize) and is removed in a washing step. Detection and relative quantification of specific genes in the DNA sample is based on the strength of the fluorescent signal that remains on the microarray after the washing step. Some microarrays use two fluorescent dyes (often red and green), which can be used to compare the range of genes present in more than one sample (Figure 6-1). Figure 6-1 shows a generic microarray analysis. DNA is isolated from groundwater collected within and outside of a contaminant groundwater plume. The DNA is labeled with two fluorescent dyes (green and red) and hybridized to the microarray. After multiple washing steps, the microarray slides are analyzed with a high-resolution laser scanner.
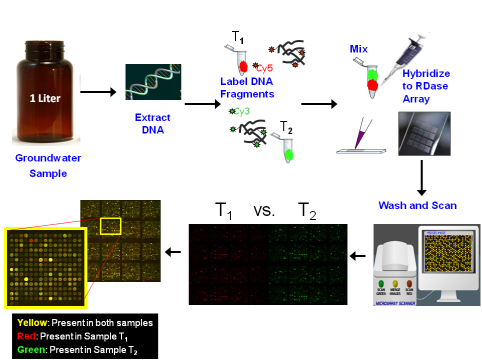
Figure 6-1. Generic microarray analysis.
Source: E. Padilla-Crespo and F. Löffler, Ph.D., University of Tennessee, 2012. Used with permission.
Microarrays can be fabricated in many forms and can contain up to hundreds of thousands of probes that target tens to thousands of genes. In high-density microarrays, the position and nucleotide sequence of each probe on the microarray is precisely known, and the fluorescence associated with each probe can be located and measured using laser excitation and detection. The benefit of high-density microarrays is that they can rapidly and simultaneously determine the presence and abundance of numerous genes in a single DNA sample using identical analytical conditions.
Microarrays have been used for environmental research for at least 15 years but have become commercially available to a wider community only recently. The commercially available Affymetrix Phylochip® microarray contains a comprehensive suite of probes for bacterial 16S rRNAA subunit of the ribosome composed of ribonucleic acid (RNA). The RNA sequence is used to classify and identify microorganisms (e.g. genus and species). genes, a stretch of DNA sequence that by convention acts as the "bar code" for bacterial identification. This microarray can therefore provide comprehensive information about microbial communityThe microorganisms present in a particular sample. structure and can answer the question “Which microorganisms are present?” (Conrad et al. 2010; Briggs et al. 2011; Cooper et al. 2011; DeAngelis et al. 2011; Mendes et al. 2011).
Another commercially available microarray, GeoChip, contains approximately 28,000 probes that target nearly 57,000 gene variants. These targeted gene variants are implicated in major microbial processes such as contaminant degradation, carbon, nitrogen, sulfur and phosphorous metabolism, metal reduction and antibiotic resistance (He et al. 2007; He et al. 2010; He et al. 2011; Zhou et al. 2011). The GeoChip thus addresses the question “What can the microbes that are present potentially do?” and, if used as an expression array for transcript analysis, can answer the question: “What are the microbes that are present actually doing?”.
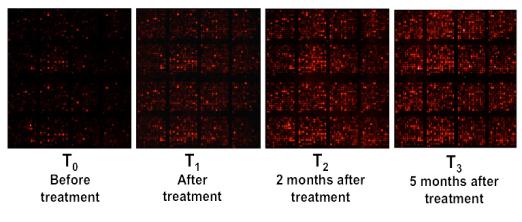
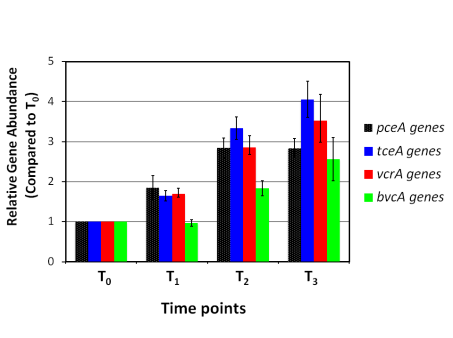
If applied to DNA samples collected over time from the same sampling locations, DNA microarrays can monitor changes in community structure and functional gene content in response to treatment such as biostimulation. Microarrays can also be used to investigate which genes are being expressed by microorganisms. This information can be used to answer questions such as “Which organisms are active?” and “What metabolic pathways are these organisms using?” These questions are addressed using expression microarrays, which require extracting mRNA from environmental samples and then converting this mRNA to complementary DNA (cDNA) in the laboratory. The cDNA is then analyzed the same way as DNA directly extracted from an environmental sample. If applied to RNA samples collected over time from the same sampling locations, expression microarrays can also monitor changes in levels of gene expression in response to treatments such as biostimulation.